

Introduction: Patients with leukemic indolent B-cell Non-Hodgkin Lymphomas (LI B-NHL) that do not easily fall into a distinct pathological category with standard diagnostics can pose a challenge as their optimal management is uncertain. Lack of a clear diagnostic label can deny patients access to novel therapies as regulatory approval of drugs is largely granted within histological categories. Moreover, these patients may be excluded from participation in clinical trials. We report findings from a prospective study evaluating targeted next generation sequencing (NGS) of circulating malignant B-cells in this setting.
Methods: Over a period of 4 years, 108 patients with LI B-NHL, deemed unclassifiable on standard peripheral blood (PB) diagnostics, were prospectively enrolled from 14 centres around the UK including ours. Patients with non-clonal lymphocytosis, confirmed CLL-type MBL, CLL (Matutes score 4 or 5), MCL, HCL, high grade lymphoma on standard PB diagnostics were excluded. Clinical and morphological characteristics were analysed. PB immunophenotyping was used for characterisation including ROR1 expression. CD15+ cells were positively selected with magnetic beads for germline DNA. Tumor-germline pairs were analysed with theEuroclonality-NGS DNA capture panel, and ARResT/Interrogate and complementary pipelines, to characterise clonal immunoglobulin rearrangements, translocations and somatic mutations (Stewart et al. ASH annual meeting 2019). Germline variants were subtracted to improve somatic mutation detection.
Results: Median age was 72yrs with M:F ratio of 2:1. Incidental lymphocytosis on a routine blood count was the commonest mode of presentation with only a third of patients symptomatic at presentation. Median lymphocyte count was 12.4 x 109/L at the time of sampling. Just under 50% of cases had partial or complete CD5 expression, CD19 and 20 expression was moderate to strong in most cases while ROR1 was negative in most cases. NGS was able to assign a diagnostic category in 11/90 (12%) cases based on detection of IGH;CCND1 (6), IGH;BCL2 (2), IGH;BCL3 (2) translocations and in one case a BRAF mutation with confirmation of hairy cell leukaemia on immuno-morphology. These tests were not triggered by standard diagnostic pathways at initial presentation. Two novel translocations of uncertain significance were detected - RAD51B:BIRC3 and IGHMswitch-MICALL2/INTS1.
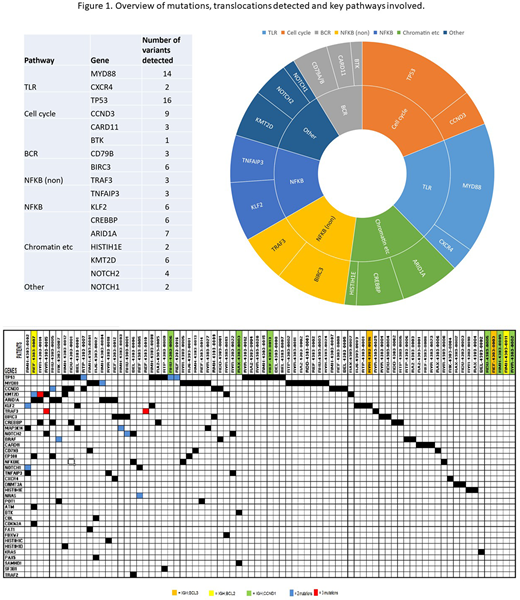
Clonal IGHV-IGHD-IGHJ rearrangements were detected by NGS and/or Sanger sequencing with predominantly IGHV3 and IGHV4 gene usage, IGHV4-34 being the most common with 15 cases. The majority of rearrangements showed somatic hypermutations above 2% and none could be assigned to the 19 major CLL-associated stereotyped subsets (using ARResT/AssignSubsets). Somatic mutations were detected in 74/108 cases. The most frequently mutated genes were TP53 in 16/108 (15%) and the hotspot MYD88 L265P mutation in 14/108 (13%) with VAF of 20-46%. Other variants detected were distributed among genes associated with NFkB signalling and chromatin remodelling (Figure 1).
The MYD88 L265P mutation was present in a fifth of cases with mutations detected (14/74) and was the sole change seen in 7 cases. Accompanying mutations in the remaining cases included CD79B (2), POT1 (1), NFKBIE (1). No concomitant CXCR4 mutations were detected. Median age of the MYD88 L265P cohort was 68 years with male predominance (M:F=10:4). 9/14 presented with an incidental lymphocytosis and median count of 11.18 x 109/L. Lymphadenopathy was present in 3/14 (21%) while 7/14 (50%) had splenomegaly. A paraprotein was detected in 6/14 cases (4 IgM, 1 IgG, 1 IgA). CD5 was expressed in 6/14 cases by flow cytometry. CD19 and 20 were uniformly positive, CD79b was variable and 12/14 cases tested did not express ROR1.
Conclusion: This prospective study outlines the value of a targeted NGS panel in enhancing the diagnosis of unclassifiable LI B-NHL thereby improving patient access to novel therapies and clinical trials. It highlights recurrent MYD88 mutations in a subset of patients that have splenomegaly as a frequent feature. 5 year clinical follow up data, currently being gathered in both the MYD88 mutated and overall cohort, will be valuable in further characterisation and risk stratification to inform management of these patients.
Furtado:Abbvie: Other: Conference Support. El-Sharkawi:Abbvie: Consultancy, Honoraria, Speakers Bureau; AstraZeneca: Consultancy, Honoraria, Speakers Bureau; Janssen: Consultancy, Speakers Bureau; Takeda: Honoraria, Speakers Bureau; Innate: Consultancy. Iyengar:Abbvie: Honoraria; Gilead: Consultancy, Honoraria, Speakers Bureau; Takeda: Consultancy, Honoraria, Speakers Bureau; Janssen: Honoraria; Beigene: Consultancy.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal